tnt
Choi et al. (2015) Nat. Biotech reanalysis: transposable elements
Sam Buckberry 29/06/2020
source("R/project_functions.R")
## Loading required package: BiocGenerics
##
## Attaching package: 'BiocGenerics'
## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs
## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, basename, cbind, colnames,
## dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
## grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
## rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
## union, unique, unsplit, which.max, which.min
## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.
## Loading required package: ggplot2
## Loading required package: lattice
## Loading required package: GenomicRanges
## Loading required package: stats4
## Loading required package: S4Vectors
##
## Attaching package: 'S4Vectors'
## The following objects are masked from 'package:base':
##
## expand.grid, I, unname
## Loading required package: IRanges
## Loading required package: GenomeInfoDb
## Loading required package: SummarizedExperiment
## Loading required package: MatrixGenerics
## Loading required package: matrixStats
##
## Attaching package: 'matrixStats'
## The following objects are masked from 'package:Biobase':
##
## anyMissing, rowMedians
##
## Attaching package: 'MatrixGenerics'
## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
## colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
## colWeightedMeans, colWeightedMedians, colWeightedSds,
## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
## rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
## rowWeightedSds, rowWeightedVars
## The following object is masked from 'package:Biobase':
##
## rowMedians
##
## Attaching package: 'magrittr'
## The following object is masked from 'package:GenomicRanges':
##
## subtract
##
## Attaching package: 'data.table'
## The following object is masked from 'package:SummarizedExperiment':
##
## shift
## The following object is masked from 'package:GenomicRanges':
##
## shift
## The following object is masked from 'package:IRanges':
##
## shift
## The following objects are masked from 'package:S4Vectors':
##
## first, second
## Loading required package: BSgenome
## Loading required package: Biostrings
## Loading required package: XVector
##
## Attaching package: 'Biostrings'
## The following object is masked from 'package:base':
##
## strsplit
## Loading required package: rtracklayer
## Loading required package: AnnotationDbi
##
## Attaching package: 'ggthemes'
## The following object is masked from 'package:cowplot':
##
## theme_map
## Loading required package: Rsamtools
##
## Attaching package: 'VariantAnnotation'
## The following object is masked from 'package:stringr':
##
## fixed
## The following object is masked from 'package:base':
##
## tabulate
##
## Attaching package: 'ChIPpeakAnno'
## The following object is masked from 'package:VariantAnnotation':
##
## info
##
## Attaching package: 'gtools'
## The following object is masked from 'package:e1071':
##
## permutations
##
## Attaching package: 'UpSetR'
## The following object is masked from 'package:lattice':
##
## histogram
## Loading required package: limma
##
## Attaching package: 'limma'
## The following object is masked from 'package:BiocGenerics':
##
## plotMA
## Loading required package: grid
##
## Attaching package: 'grid'
## The following object is masked from 'package:Biostrings':
##
## pattern
TE quantification (not run)
parallel -j 35 ~/working_data_01/bin/TElocal/TElocal -b {} \
--GTF /home/sbuckberry/working_data_02/polo_project/human_ips/resources/hg19_ensGene_with_ercc.gtf \
--TE /home/sbuckberry/working_data_02/polo_project/human_ips/resources/hg19_rmsk_TElocus.ind \
--project {.} \
--format BAM \
--sortByPos \
--stranded reverse \
--mode uniq ::: SRS*uniq.bam
Read the sample count tables and combine into matrix
# # List the counts tables for all samples
# cnt_files <- list.files(path = "RNAseq/hochedlinger_data/te_analysis/", full.names = TRUE, pattern = "cntTable.gz")
#
# dat2 <- lapply(cnt_files, read.table, header = TRUE, row.names = 1) %>% do.call(cbind, .)
# libs <- colnames(dat2) %>% str_sub(start = 1, end = 10)
# colnames(dat2) <- libs
#
# #remove rows with all zero's
# keep_row <- rowSums(dat2) > 0
# table(keep_row)
# dat2 <- dat2[keep_row, ]
# saveRDS(dat2, file = "RNAseq/hochedlinger_data/all_te_and_gene_counts.Rds")
dat2 <- readRDS("RNAseq/hochedlinger_data/all_te_and_gene_counts.Rds")
dim(dat2)
## [1] 2048776 63
Read the meta-data table and match to expression data
sample_dat <- fread("RNAseq/hochedlinger_data/sample_sheet.csv")
sample_dat <- sample_dat[match(colnames(dat2), sample_dat$secondary_sample_accession), ]
all(colnames(dat2) == sample_dat$Run)
## [1] TRUE
Read in the count data
y2 <- DGEList(counts = dat2)
y2$samples <- cbind(y2$samples, sample_dat)
Load the transposable element data
## Get the TE genome co-ordinates
repeat_gtf <- read.table("resources/hg19_rmsk_TE.gtf.gz")
repeat_gr <- GRanges(seqnames = repeat_gtf$V1,
ranges = IRanges(start = repeat_gtf$V4,
end = repeat_gtf$V5))
strand(repeat_gr) <- repeat_gtf$V7
repeat_gr$class <- repeat_gtf$V19
repeat_gr$family <- repeat_gtf$V16
repeat_gr$gene <- repeat_gtf$V10
repeat_gr$transcript <- repeat_gtf$V13
repeat_gr$id <- str_c(repeat_gr$transcript, repeat_gr$family, repeat_gr$class, sep = ":")
rm(repeat_gtf)
saveRDS(repeat_gr, "resources/hg19_rmsk_TE_granges.Rds")
repeat_gr <- readRDS("resources/hg19_rmsk_TE_granges.Rds")
repeat_gr <- readRDS("resources/hg19_rmsk_TE_granges.Rds")
Function to test DE for each genetic background
test_te_de <- function(background="HUES3", groups=c("iPSC", "ESC_GFP"), dataset="data set: isogenic"){
y_sub <- y2[ ,(y2$samples$Background %in% background) &
(y2$samples$Group %in% groups) &
(y2$samples$characteristics_ch1 == dataset)]
dim(y_sub)
design <- model.matrix(~Group, data=y_sub$samples)
colnames(design) <- str_remove(string = colnames(design), pattern = "Group")
keep <- filterByExpr(y_sub, design)
table(keep)
y_sub <- y_sub[keep, ,keep.lib.sizes=FALSE]
y_sub <- calcNormFactors(y_sub)
y_sub <- estimateDisp(y_sub, design = design, robust = TRUE)
plotBCV(y_sub)
plotMDS(y_sub[grepl(pattern = "_dup", rownames(y_sub)), ], labels = y_sub$samples$source_name_ch1)
fit <- glmFit(y_sub, design)
lrt <- glmLRT(fit, contrast=c(0,-1))
tt <- topTags(lrt, n = nrow(y_sub))
tt_table <- tt$table
tt_table$gene_id <- rownames(tt_table)
tt_table$contrast <- background
# Subset for TE's and recalculate FDR
tt_table <- tt_table[tt_table$gene_id %in% repeat_gr$id, ]
tt_table$FDR <- p.adjust(p = tt_table$PValue, method = "fdr")
tt_table$significant <- (abs(tt_table$logFC) > 1) & (tt_table$FDR < 0.05) & (tt_table$logCPM > 0)
tt_table$significant <- ifelse(test = tt_table$significant, yes = "Significant", no = "NS")
tt_table$significant <- factor(tt_table$significant, levels = c("NS", "Significant"))
# Add the TE locus information
ind <- match(tt_table$gene_id, repeat_gr$id)
tt_table <- cbind(tt_table, as.data.frame(repeat_gr)[ind, ])
}
hues3_tt <- test_te_de(background = "HUES3")
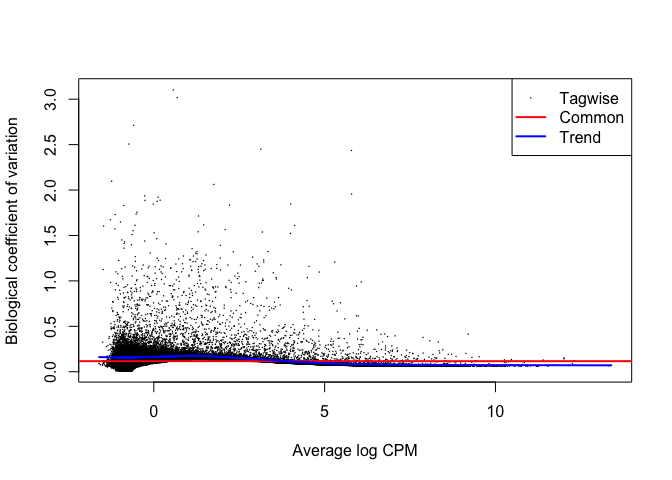
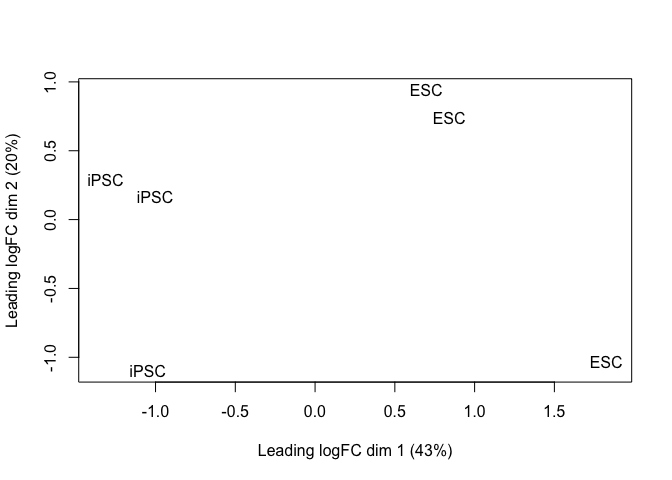
hues2_tt <- test_te_de(background = "HUES2")
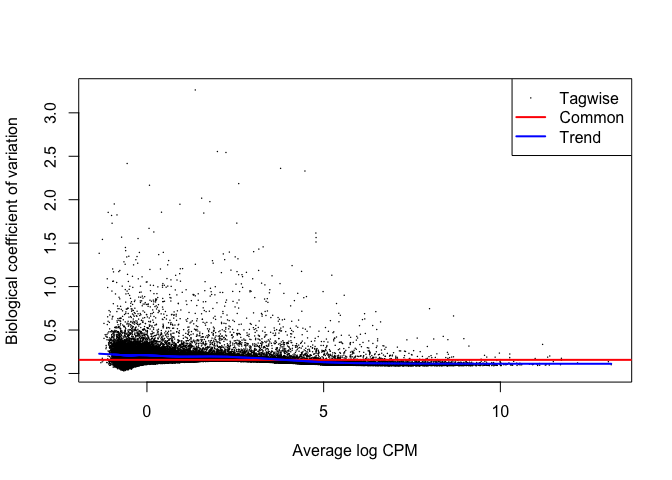
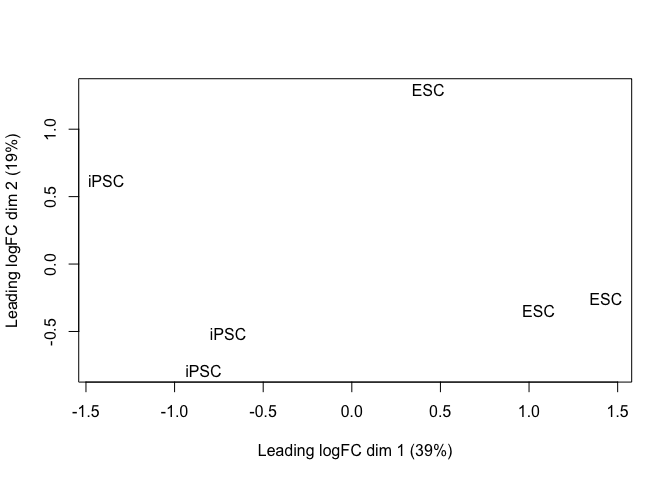
hues2_GFP_tt <- test_te_de(background = "HUES2", groups = c("ESC", "ESC_GFP"))
all_tt <- rbind(hues2_tt, hues3_tt)
te_de_df <- all_tt
sig_ids <- te_de_df$gene_id[te_de_df$significant == "Significant"] %>% unique()
background <- c("HUES2", "HUES3")
groups <- c("iPSC", "ESC_GFP")
dataset <- "data set: isogenic"
y_sub <- y2[ ,(y2$samples$Background %in% background) &
(y2$samples$Group %in% groups) &
(y2$samples$characteristics_ch1 == dataset)]
all_cpm <- log2(cpm(y_sub) + 1)
hm_annot_dat <- y_sub$samples[ ,c("Background", "Group")]
col_ids <- str_c(y_sub$samples$title)
plot_dat <- all_cpm[rownames(all_cpm) %in% sig_ids, ]
#plot_dat <- all_cpm[grepl("HERVH-int", rownames(all_cpm)),
# y_sub$samples$source_name_ch1 != "Fibroblast"]
plot_dat <- plot_dat[complete.cases(plot_dat), ] %>% data.frame()
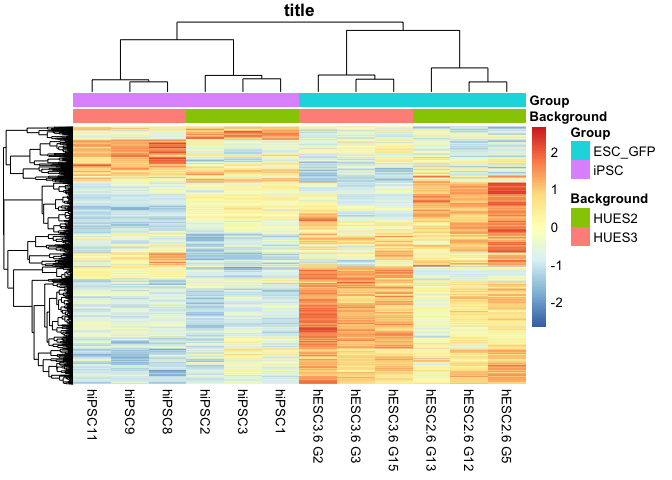
pdf("RNAseq/plots/Choi_TE_heatmap_combined.pdf", width = 4, height = 5)
pheatmap(plot_dat, scale = 'row',
annotation_col = hm_annot_dat,
labels_col = col_ids, main = "title",
annotation_names_col = TRUE,
show_rownames = FALSE, border_color = NA,
clustering_distance_rows = 'correlation',
clustering_distance_cols = 'correlation')
dev.off()
## pdf
## 3
pheatmap(plot_dat, scale = 'row',
annotation_col = hm_annot_dat,
labels_col = col_ids, main = "title",
annotation_names_col = TRUE,
show_rownames = FALSE, border_color = NA,
clustering_distance_rows = 'correlation',
clustering_distance_cols = 'correlation')
wb_ed_fig8k <- openxlsx::createWorkbook()
openxlsx::addWorksheet(wb_ed_fig8k, sheetName = "ED_Fig_8k")
openxlsx::writeData(wb = wb_ed_fig8k, sheet = "ED_Fig_8k",
x = plot_dat)
openxlsx::saveWorkbook(wb = wb_ed_fig8k,
file = "ED_Figure_8k_source_data.xlsx", overwrite = TRUE)