tnt
Extended Data Figures 3g,h,i: Ma et al CG DMR reanalysis
Sam Buckberry 2023-07-13
source("R/project_functions.R")
This section was run on remote server and code is not executed here
### Call iPSC vs ESC DMRs
library(bsseq)
library(dmrseq)
library(magrittr)
library(stringr)
library(BiocParallel)
library(data.table)
obj_files <- c("SRS606777_CG_bsseq_obj.Rds",
"SRS606778_CG_bsseq_obj.Rds",
"SRS606782_CG_bsseq_obj.Rds",
"SRS606783_CG_bsseq_obj.Rds",
"SRS606784_CG_bsseq_obj.Rds",
"SRS606785_CG_bsseq_obj.Rds")
obj_files <- str_c("/home/sbuckberry/working_data_02/polo_project/human_ips/methylCseq/processed_data/bsseq_objects/bsobj_CG/", obj_files)
#obj_files <- str_c("~/Datasets/human_data/bsobj_CG/", obj_files)
# Read a Bs_seq boject from .Rds file
read_bs_obj <- function(rds_path){
bs_obj <- readRDS(file = rds_path)
bs_obj <- strandCollapse(bs_obj)
return(bs_obj)
}
#--------Call DMRs
# Load the data
obj_list <- lapply(X = obj_files, read_bs_obj)
obj_list <- bsseq::combineList(x = obj_list)
pData(obj_list)$CellType <- factor(c(rep("ESC", times=2), rep("iPSC", times=4)))
pData(obj_list)$Replicate <- c(1:2,1:4)
# Remove CpG with no coverage
loci.idx <- which(DelayedMatrixStats::rowSums2(getCoverage(obj_list, type="Cov")==0) == 0)
obj_list <- obj_list[loci.idx, ]
# Save object for easy retreval
saveRDS(object = obj_list,
file = "/home/sbuckberry/working_data_02/polo_project/human_ips/methylCseq/processed_data/dmrseq_dmrs/Mitalipov_BSseq_obj.rds")
BiocParallel::register(BiocParallel::SerialParam())
mitalipov_dmrs <- dmrseq(obj_list, testCovariate = "CellType",
bpSpan = 500,
maxGap = 500,
maxPerms = 10,
chrsPerChunk = 1)
saveRDS(object = mitalipov_dmrs,
file = "/home/sbuckberry/working_data_02/polo_project/human_ips/methylCseq/processed_data/dmrseq_dmrs/Mitalipov_dmrs.Rds")
Calculate mCG/CG delta and SCNT correction
gr_dmr <- readRDS("wgbs/dmrs/dmr_out/Mitalipov_dmrs.Rds")
obj_files <- list.files("wgbs/CG_bsobj/", pattern = "SRS6067", full.names = TRUE)
test_fls <- obj_files[basename(obj_files) %in% c("SRS606777_CG_bsseq_obj.Rds",
"SRS606778_CG_bsseq_obj.Rds",
"SRS606782_CG_bsseq_obj.Rds",
"SRS606783_CG_bsseq_obj.Rds",
"SRS606784_CG_bsseq_obj.Rds",
"SRS606785_CG_bsseq_obj.Rds")]
groups <- c(rep("ESC", times=2), rep("iPSC", times=4))
filter_dmrs <- function(gr_dmr, obj_fls, groups, cores=1){
### Check number of groups = 2
stopifnot(length(unique(groups)) == 2)
group1 <- unique(groups)[1]
group2 <- unique(groups)[2]
# Get the DMR sequence and the number of CG positions in DMRs
seqs <- getSeq(BSgenome.Hsapiens.UCSC.hg19, gr_dmr)
CpG_count <- dinucleotideFrequency(seqs)[ ,"CG"]
rm(seqs)
# Calculate mCG and CG totals for DMRs
mCG_levels <- make_mC_matrix(obj_fls = obj_fls, gr = gr_dmr, cores = cores)
mCG_levels <- data.frame(mCG_levels)
# Calculate mean delta and variance between groups
group1_mean <- apply(mCG_levels[ ,group1 == groups], MARGIN = 1, mean, na.rm=TRUE)
group1_sd <- apply(mCG_levels[ ,group1 == groups], MARGIN = 1, sd, na.rm=TRUE)
group2_mean <- apply(mCG_levels[ ,group2 == groups], MARGIN = 1, mean, na.rm=TRUE)
group2_sd <- apply(mCG_levels[ ,group2 == groups], MARGIN = 1, sd, na.rm=TRUE)
mCG_delta <- group1_mean - group2_mean
# Check dimensions
stopifnot(length(gr_dmr) == nrow(mCG_levels))
# Add all data to GRanges
dat <- cbind(mCG_delta, CpG_count, data.frame(mCG_levels))
# Round the metrics
dat <- round(dat, digits = 3)
# Add the metadata columns to the gr object
mcols(gr_dmr) <- cbind(mcols(gr_dmr), dat)
return(gr_dmr)
}
gr_dmr_annotated <- filter_dmrs(gr_dmr = gr_dmr, obj_fls = test_fls,
groups = groups, cores = 1)
## Reading wgbs/CG_bsobj//SRS606777_CG_bsseq_obj.Rds 2023-07-13 16:55:15
## Reading wgbs/CG_bsobj//SRS606777_CG_bsseq_obj.Rds
## 2023-07-13 16:55:15
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606778_CG_bsseq_obj.Rds 2023-07-13 16:55:26
## Reading wgbs/CG_bsobj//SRS606778_CG_bsseq_obj.Rds
## 2023-07-13 16:55:26
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606782_CG_bsseq_obj.Rds 2023-07-13 16:55:35
## Reading wgbs/CG_bsobj//SRS606782_CG_bsseq_obj.Rds
## 2023-07-13 16:55:35
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606783_CG_bsseq_obj.Rds 2023-07-13 16:55:42
## Reading wgbs/CG_bsobj//SRS606783_CG_bsseq_obj.Rds
## 2023-07-13 16:55:42
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606784_CG_bsseq_obj.Rds 2023-07-13 16:55:49
## Reading wgbs/CG_bsobj//SRS606784_CG_bsseq_obj.Rds
## 2023-07-13 16:55:49
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606785_CG_bsseq_obj.Rds 2023-07-13 16:55:56
## Reading wgbs/CG_bsobj//SRS606785_CG_bsseq_obj.Rds
## 2023-07-13 16:55:56
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Making matrix of mC levels for regions...
ips_esc_dmrs_heso <- gr_dmr_annotated[(abs(gr_dmr_annotated$mCG_delta) > 0.20) & (gr_dmr_annotated$pval < 0.05)]
# Count overlaps with donor DMRs
#all_dmrs <- readRDS("methylCseq/processed_data/dmrseq_dmrs/classified_dmrs_granges.Rds")
#table(overlapsAny(ips_esc_dmrs_heso, all_dmrs))
Profile DMR correction using the same method as primary donor samples
### Calc mCG/CG in DMRs for all samples
ips_esc_dmrs_mCG <- make_mC_matrix(obj_fls = obj_files, gr = ips_esc_dmrs_heso, cores = 1)
## Reading wgbs/CG_bsobj//SRS606775_CG_bsseq_obj.Rds 2023-07-13 16:56:07
## Reading wgbs/CG_bsobj//SRS606775_CG_bsseq_obj.Rds
## 2023-07-13 16:56:07
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606776_CG_bsseq_obj.Rds 2023-07-13 16:56:13
## Reading wgbs/CG_bsobj//SRS606776_CG_bsseq_obj.Rds
## 2023-07-13 16:56:13
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606777_CG_bsseq_obj.Rds 2023-07-13 16:56:18
## Reading wgbs/CG_bsobj//SRS606777_CG_bsseq_obj.Rds
## 2023-07-13 16:56:18
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606778_CG_bsseq_obj.Rds 2023-07-13 16:56:24
## Reading wgbs/CG_bsobj//SRS606778_CG_bsseq_obj.Rds
## 2023-07-13 16:56:24
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606779_CG_bsseq_obj.Rds 2023-07-13 16:56:30
## Reading wgbs/CG_bsobj//SRS606779_CG_bsseq_obj.Rds
## 2023-07-13 16:56:30
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606780_CG_bsseq_obj.Rds 2023-07-13 16:56:36
## Reading wgbs/CG_bsobj//SRS606780_CG_bsseq_obj.Rds
## 2023-07-13 16:56:36
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606781_CG_bsseq_obj.Rds 2023-07-13 16:56:42
## Reading wgbs/CG_bsobj//SRS606781_CG_bsseq_obj.Rds
## 2023-07-13 16:56:42
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606782_CG_bsseq_obj.Rds 2023-07-13 16:56:47
## Reading wgbs/CG_bsobj//SRS606782_CG_bsseq_obj.Rds
## 2023-07-13 16:56:47
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606783_CG_bsseq_obj.Rds 2023-07-13 16:56:53
## Reading wgbs/CG_bsobj//SRS606783_CG_bsseq_obj.Rds
## 2023-07-13 16:56:53
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606784_CG_bsseq_obj.Rds 2023-07-13 16:56:59
## Reading wgbs/CG_bsobj//SRS606784_CG_bsseq_obj.Rds
## 2023-07-13 16:56:59
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Reading wgbs/CG_bsobj//SRS606785_CG_bsseq_obj.Rds 2023-07-13 16:57:05
## Reading wgbs/CG_bsobj//SRS606785_CG_bsseq_obj.Rds
## 2023-07-13 16:57:05
## Calculating coverage...
## Calculating M...
## Calculating methylation percentage...
## Making matrix of mC levels for regions...
mcols(ips_esc_dmrs_heso) <- cbind(mcols(ips_esc_dmrs_heso), ips_esc_dmrs_mCG)
colnames(ips_esc_dmrs_mCG)
## [1] "SRS606775_CG_bsseq_obj.Rds" "SRS606776_CG_bsseq_obj.Rds"
## [3] "SRS606777_CG_bsseq_obj.Rds" "SRS606778_CG_bsseq_obj.Rds"
## [5] "SRS606779_CG_bsseq_obj.Rds" "SRS606780_CG_bsseq_obj.Rds"
## [7] "SRS606781_CG_bsseq_obj.Rds" "SRS606782_CG_bsseq_obj.Rds"
## [9] "SRS606783_CG_bsseq_obj.Rds" "SRS606784_CG_bsseq_obj.Rds"
## [11] "SRS606785_CG_bsseq_obj.Rds"
esc_id <- c("SRS606777", "SRS606778")
ips_id <- c("SRS606782", "SRS606783", "SRS606784", "SRS606785")
fib_id <- c("SRS606776")
scnt_id <- c("SRS606775", "SRS606779", "SRS606780", "SRS606781")
colnames(ips_esc_dmrs_mCG) <- str_remove(string = colnames(ips_esc_dmrs_mCG),
pattern = "_CG_bsseq_obj.Rds")
esc_ips_delta <- rowMeans(ips_esc_dmrs_mCG[ ,ips_id], na.rm = TRUE) - rowMeans(ips_esc_dmrs_mCG[ ,esc_id], na.rm = TRUE)
fib_ips_delta <- rowMeans(ips_esc_dmrs_mCG[ ,ips_id], na.rm = TRUE) - ips_esc_dmrs_mCG[ ,fib_id]
esc_scnt_delta <- rowMeans(ips_esc_dmrs_mCG[ ,scnt_id], na.rm = TRUE) - rowMeans(ips_esc_dmrs_mCG[ ,esc_id], na.rm = TRUE)
dmr_delta_df <- data.frame(ESC_delta = esc_ips_delta, Fibroblast_delta = fib_ips_delta)
dmr_delta_df$dmr <- NA
dmr_delta_df$dmr[dmr_delta_df$ESC_delta > 0.2 & dmr_delta_df$Fibroblast_delta >= 0.2] <- "Aberrant_hypermethylation"
dmr_delta_df$dmr[dmr_delta_df$ESC_delta < -0.2 & dmr_delta_df$Fibroblast_delta <= -0.2] <- "Aberrant_hypomethylation"
dmr_delta_df$dmr[dmr_delta_df$ESC_delta < -0.2 & abs(dmr_delta_df$Fibroblast_delta) <= 0.2 ] <- "Memory_hypomethylation"
dmr_delta_df$dmr[dmr_delta_df$ESC_delta > 0.2 & abs(dmr_delta_df$Fibroblast_delta) <= 0.2 ] <- "Memory_hypermethylation"
dmr_delta_df$dmr[dmr_delta_df$ESC_delta < -0.2 & dmr_delta_df$Fibroblast_delta >= 0.2] <- "Partial_hypomethylation_memory"
dmr_delta_df$dmr[dmr_delta_df$ESC_delta > 0.2 & dmr_delta_df$Fibroblast_delta <= -0.2] <- "Partial_hypermethyation_memory"
dmr_delta_df$dmr <- factor(dmr_delta_df$dmr)
table(dmr_delta_df$dmr)
##
## Aberrant_hypermethylation Aberrant_hypomethylation
## 1446 105
## Memory_hypermethylation Memory_hypomethylation
## 505 719
## Partial_hypermethyation_memory Partial_hypomethylation_memory
## 30 153
table(dmr_delta_df$Class)
## < table of extent 0 >
table(dmr_delta_df$dmr) %>% sum()
## [1] 2958
dmr_delta_df$Class <- ifelse(test = dmr_delta_df$ESC_delta < 0, yes = "Hypo", no = "Hyper")
dmr_delta_df$Class <- factor(dmr_delta_df$Class, levels=c("Hyper", "Hypo"))
all(rownames(dmr_delta_df) == gr_to_loci(ips_esc_dmrs_heso))
## [1] TRUE
ips_esc_dmrs_heso$class <- dmr_delta_df$dmr
ips_esc_dmrs_heso$direction <- dmr_delta_df$Class
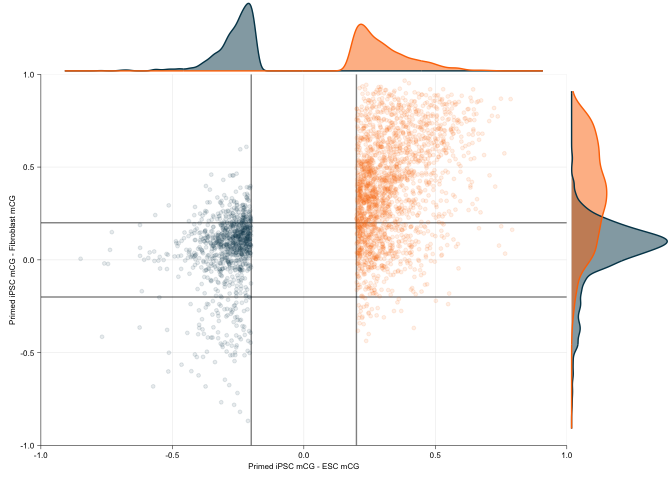
dmr_scatter_gg <- ggplot(data = dmr_delta_df, mapping = aes(x = ESC_delta, y = Fibroblast_delta, colour = Class, fill = Class)) +
geom_vline(xintercept = c(-.2, .2), alpha=0.5) + geom_hline(yintercept = c(-.2, .2), alpha=0.5) +
geom_point(alpha=0.1, inherit.aes = TRUE, size=1) +
scale_x_continuous(limits = c(-1, 1), expand = c(0,0)) + scale_y_continuous(limits = c(-1, 1), expand = c(0,0)) +
ylab("Primed iPSC mCG - Fibroblast mCG") +
xlab("Primed iPSC mCG - ESC mCG") +
scale_colour_manual(values = vitC[c(6,1)]) +
#scale_colour_manual(values = brewer.pal(n = 2, name = "Dark2")) +
sams_pub_theme(x.text.angle = 0, hjust = 0.5)
## Warning: The `size` argument of `element_line()` is deprecated as of ggplot2 3.4.0.
## ℹ Please use the `linewidth` argument instead.
dmr_scatter_gg <- ggExtra::ggMarginal(dmr_scatter_gg, groupColour = TRUE, groupFill=TRUE)
dmr_scatter_gg
Plot the correction of the SCNT cells as histograms
esc_id <- c("SRS606777", "SRS606778")
sendai_id <- c("SRS606782", "SRS606783")
retro_id <- c("SRS606784", "SRS606785")
fib_id <- c("SRS606776")
scnt_id <- c("SRS606775", "SRS606779", "SRS606780", "SRS606781")
sendai_delta <- rowMeans(ips_esc_dmrs_mCG[ ,sendai_id], na.rm = TRUE) - rowMeans(ips_esc_dmrs_mCG[ ,esc_id], na.rm = TRUE)
retro_delta <- rowMeans(ips_esc_dmrs_mCG[ ,retro_id], na.rm = TRUE) - rowMeans(ips_esc_dmrs_mCG[ ,esc_id], na.rm = TRUE)
scnt_delta <- rowMeans(ips_esc_dmrs_mCG[ ,scnt_id], na.rm = TRUE) - rowMeans(ips_esc_dmrs_mCG[ ,esc_id], na.rm = TRUE)
fib_ips_delta <- rowMeans(ips_esc_dmrs_mCG[ ,c(sendai_id, retro_id)], na.rm = TRUE) - ips_esc_dmrs_mCG[ ,fib_id]
esc_ips_delta <- rowMeans(ips_esc_dmrs_mCG[ ,c(sendai_id, retro_id)], na.rm = TRUE) - rowMeans(ips_esc_dmrs_mCG[ ,esc_id], na.rm = TRUE)
dmr_delta_df <- data.frame(ESC_delta = esc_ips_delta, Fibroblast_delta = fib_ips_delta, SCNT_delta = scnt_delta)
dmr_delta_df$Class <- ifelse(test = dmr_delta_df$ESC_delta < 0, yes = "Hypo", no = "Hyper")
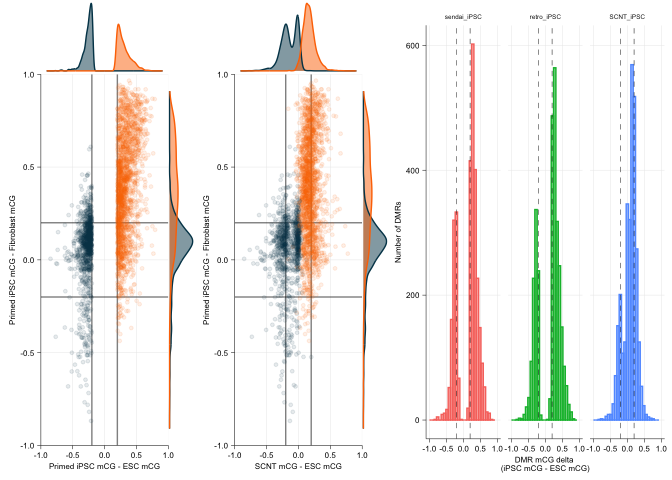
ipsc_scatter <- ggplot(data = dmr_delta_df, mapping = aes(x = ESC_delta, y = Fibroblast_delta, colour = Class, fill = Class)) +
geom_vline(xintercept = c(-.2, .2), alpha=0.5) + geom_hline(yintercept = c(-.2, .2), alpha=0.5) +
geom_point(alpha=0.1, inherit.aes = TRUE, size=1) +
scale_x_continuous(limits = c(-1, 1), expand = c(0,0)) + scale_y_continuous(limits = c(-1, 1), expand = c(0,0)) +
ylab("Primed iPSC mCG - Fibroblast mCG") +
xlab("Primed iPSC mCG - ESC mCG") +
scale_colour_manual(values = vitC[c(6,1)]) +
#scale_colour_manual(values = brewer.pal(n = 2, name = "Dark2")) +
sams_pub_theme(x.text.angle = 0, hjust = 0.5)
ipsc_scatter_gg <- ggExtra::ggMarginal(ipsc_scatter, groupColour = TRUE, groupFill=TRUE)
scnt_scatter <- ggplot(data = dmr_delta_df, mapping = aes(x = SCNT_delta, y = Fibroblast_delta, colour = Class, fill = Class)) +
geom_vline(xintercept = c(-.2, .2), alpha=0.5) + geom_hline(yintercept = c(-.2, .2), alpha=0.5) +
geom_point(alpha=0.1, inherit.aes = TRUE, size=1) +
scale_x_continuous(limits = c(-1, 1), expand = c(0,0)) + scale_y_continuous(limits = c(-1, 1), expand = c(0,0)) +
ylab("Primed iPSC mCG - Fibroblast mCG") +
xlab("SCNT mCG - ESC mCG") +
scale_colour_manual(values = vitC[c(6,1)]) +
#scale_colour_manual(values = brewer.pal(n = 2, name = "Dark2")) +
sams_pub_theme(x.text.angle = 0, hjust = 0.5)
scnt_scatter_gg <- ggExtra::ggMarginal(scnt_scatter, groupColour = TRUE, groupFill=TRUE)
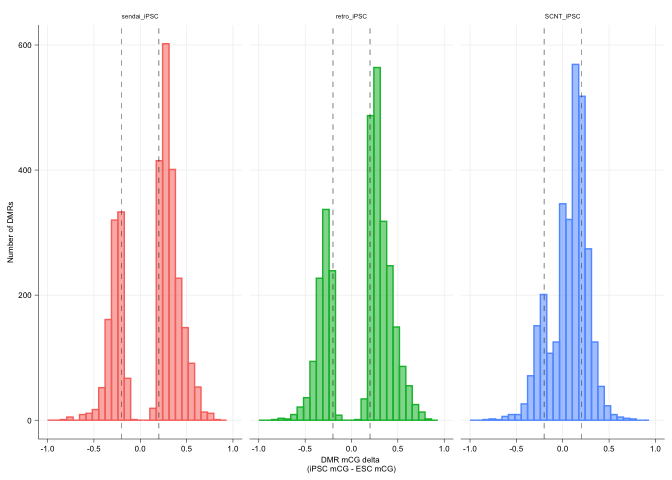
delta_df <- data.frame(sendai_iPSC=sendai_delta,
retro_iPSC=retro_delta,
SCNT_iPSC=scnt_delta)
delta_df_melt <- reshape2::melt(delta_df)
## No id variables; using all as measure variables
line_mm <- 0.5 / 2.835
gg_delta_correct <- ggplot(delta_df_melt,
aes(x = value, group=variable,
color=variable,
fill=variable, alpha=0.9)) +
geom_histogram(bins = 30, position="identity") +
geom_vline(xintercept = c(-.2, .2), linetype='dashed', size = line_mm) +
#geom_density() +
#scale_color_manual(values=c(reprog_pal[1:2], vitC[c(2,2)])) +
#scale_fill_manual(values=c(reprog_pal[1:2], vitC[c(2,2)])) +
facet_grid(.~variable, scales = "free_y") +
scale_x_continuous(limits = c(-1, 1)) +
xlab("DMR mCG delta \n(iPSC mCG - ESC mCG)") +
ylab("Number of DMRs") +
sams_pub_theme(x.text.angle = 0, hjust = 0.5)
## Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
## ℹ Please use `linewidth` instead.
gg_delta_correct
## Warning: Removed 6 rows containing missing values (`geom_bar()`).
pdf("wgbs/plots/ma_cell_line_dmr_correction_plots.pdf", width = 7, height = 2)
plot_grid(ipsc_scatter_gg, scnt_scatter_gg, gg_delta_correct, ncol = 3, nrow = 1,
rel_widths = c(0.75, 0.75, 1.1))
## Warning: Removed 6 rows containing missing values (`geom_bar()`).
dev.off()
## quartz_off_screen
## 2
png("wgbs/plots/ma_cell_line_dmr_correction_plots.png", width = 7, height = 2, units = "in", res = 300)
plot_grid(ipsc_scatter_gg, scnt_scatter_gg, gg_delta_correct, ncol = 3, nrow = 1,
rel_widths = c(0.75, 0.75, 1.1))
## Warning: Removed 6 rows containing missing values (`geom_bar()`).
dev.off()
## quartz_off_screen
## 2
plot_grid(ipsc_scatter_gg, scnt_scatter_gg, gg_delta_correct, ncol = 3, nrow = 1,
rel_widths = c(0.75, 0.75, 1.1))
## Warning: Removed 6 rows containing missing values (`geom_bar()`).
Barplot of correction with respect to CH-DMRs
delta_df_melt$delta_pass <- abs(delta_df_melt$value) < 0.2
dmr_rate <- delta_df_melt %>% dplyr::group_by(variable, delta_pass) %>% dplyr::count()
gg_bar <- ggplot(dmr_rate[dmr_rate$variable == "SCNT_iPSC", ],
aes(x = variable, y = n, fill=delta_pass)) +
geom_bar(stat = 'identity') +
sams_pub_theme()
pdf("wgbs/plots/ma_scnt_dmr_correction_fraction_barplot.pdf", width = 1, height = 2)
gg_bar
dev.off()
## quartz_off_screen
## 2
gg_bar
Export source data for manuscript
wb_ed_fig_3ghi <- openxlsx::createWorkbook()
openxlsx::addWorksheet(wb_ed_fig_3ghi, sheetName = "ED_Fig_3g")
openxlsx::writeData(wb = wb_ed_fig_3ghi, sheet = "ED_Fig_3g",
x = gg_bar$data)
openxlsx::addWorksheet(wb_ed_fig_3ghi, sheetName = "ED_Fig_3h")
openxlsx::writeData(wb = wb_ed_fig_3ghi, sheet = "ED_Fig_3h",
x = rbind(ipsc_scatter$data, scnt_scatter$data))
openxlsx::addWorksheet(wb_ed_fig_3ghi, sheetName = "ED_Fig_3i")
openxlsx::writeData(wb = wb_ed_fig_3ghi, sheet = "ED_Fig_3i",
x = gg_delta_correct$data)
openxlsx::saveWorkbook(wb = wb_ed_fig_3ghi,
file = "ED_Figure_3ghi_source_data.xlsx",
overwrite = TRUE)
sessionInfo()
## R version 4.2.1 (2022-06-23)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur ... 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_AU.UTF-8/en_AU.UTF-8/en_AU.UTF-8/C/en_AU.UTF-8/en_AU.UTF-8
##
## attached base packages:
## [1] grid parallel stats4 stats graphics grDevices utils
## [8] datasets methods base
##
## other attached packages:
## [1] RColorBrewer_1.1-3
## [2] XML_3.99-0.12
## [3] ggExtra_0.10.0
## [4] gprofiler2_0.2.1
## [5] gt_0.8.0
## [6] Gviz_1.40.1
## [7] edgeR_3.38.4
## [8] limma_3.52.4
## [9] UpSetR_1.4.0
## [10] gtools_3.9.4
## [11] ggdendro_0.1.23
## [12] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
## [13] ChIPpeakAnno_3.30.1
## [14] ggridges_0.5.4
## [15] ggalluvial_0.12.3
## [16] alluvial_0.1-2
## [17] VariantAnnotation_1.42.1
## [18] Rsamtools_2.12.0
## [19] ggthemes_4.2.4
## [20] cowplot_1.1.1
## [21] ggrepel_0.9.2
## [22] ggfortify_0.4.15
## [23] pheatmap_1.0.12
## [24] GenomicFeatures_1.48.4
## [25] AnnotationDbi_1.58.0
## [26] BSgenome.Hsapiens.UCSC.hg19_1.4.3
## [27] BSgenome_1.64.0
## [28] rtracklayer_1.56.1
## [29] Biostrings_2.64.1
## [30] XVector_0.36.0
## [31] data.table_1.14.6
## [32] readxl_1.4.1
## [33] openxlsx_4.2.5.1
## [34] stringr_1.5.0
## [35] magrittr_2.0.3
## [36] bsseq_1.32.0
## [37] SummarizedExperiment_1.26.1
## [38] MatrixGenerics_1.8.1
## [39] matrixStats_0.63.0
## [40] GenomicRanges_1.48.0
## [41] GenomeInfoDb_1.32.4
## [42] IRanges_2.30.1
## [43] S4Vectors_0.34.0
## [44] e1071_1.7-12
## [45] caret_6.0-93
## [46] lattice_0.20-45
## [47] ggplot2_3.4.1
## [48] Biobase_2.56.0
## [49] BiocGenerics_0.42.0
## [50] preprocessCore_1.58.0
##
## loaded via a namespace (and not attached):
## [1] rappdirs_0.3.3 ModelMetrics_1.2.2.2
## [3] R.methodsS3_1.8.2 tidyr_1.2.1
## [5] bit64_4.0.5 knitr_1.41
## [7] DelayedArray_0.22.0 R.utils_2.12.2
## [9] rpart_4.1.19 KEGGREST_1.36.3
## [11] hardhat_1.2.0 RCurl_1.98-1.9
## [13] AnnotationFilter_1.20.0 generics_0.1.3
## [15] lambda.r_1.2.4 RSQLite_2.2.19
## [17] proxy_0.4-27 future_1.29.0
## [19] bit_4.0.5 xml2_1.3.3
## [21] lubridate_1.9.0 httpuv_1.6.6
## [23] assertthat_0.2.1 gower_1.0.0
## [25] xfun_0.35 hms_1.1.2
## [27] evaluate_0.18 promises_1.2.0.1
## [29] fansi_1.0.4 restfulr_0.0.15
## [31] progress_1.2.2 dbplyr_2.2.1
## [33] DBI_1.1.3 htmlwidgets_1.5.4
## [35] futile.logger_1.4.3 purrr_0.3.5
## [37] ellipsis_0.3.2 dplyr_1.0.10
## [39] backports_1.4.1 permute_0.9-7
## [41] biomaRt_2.52.0 deldir_1.0-6
## [43] sparseMatrixStats_1.8.0 vctrs_0.5.2
## [45] ensembldb_2.20.2 cachem_1.0.6
## [47] withr_2.5.0 checkmate_2.1.0
## [49] GenomicAlignments_1.32.1 prettyunits_1.1.1
## [51] cluster_2.1.4 lazyeval_0.2.2
## [53] crayon_1.5.2 labeling_0.4.2
## [55] recipes_1.0.3 pkgconfig_2.0.3
## [57] nlme_3.1-160 ProtGenerics_1.28.0
## [59] nnet_7.3-18 rlang_1.0.6
## [61] globals_0.16.2 lifecycle_1.0.3
## [63] miniUI_0.1.1.1 filelock_1.0.2
## [65] BiocFileCache_2.4.0 dichromat_2.0-0.1
## [67] VennDiagram_1.7.3 cellranger_1.1.0
## [69] graph_1.74.0 Matrix_1.5-3
## [71] Rhdf5lib_1.18.2 base64enc_0.1-3
## [73] png_0.1-8 viridisLite_0.4.1
## [75] rjson_0.2.21 bitops_1.0-7
## [77] R.oo_1.25.0 rhdf5filters_1.8.0
## [79] pROC_1.18.0 blob_1.2.3
## [81] DelayedMatrixStats_1.18.2 regioneR_1.28.0
## [83] parallelly_1.32.1 jpeg_0.1-10
## [85] scales_1.2.1 memoise_2.0.1
## [87] plyr_1.8.8 zlibbioc_1.42.0
## [89] compiler_4.2.1 BiocIO_1.6.0
## [91] cli_3.6.0 listenv_0.8.0
## [93] htmlTable_2.4.1 formatR_1.12
## [95] Formula_1.2-4 MASS_7.3-58.1
## [97] tidyselect_1.2.0 stringi_1.7.12
## [99] highr_0.9 yaml_2.3.6
## [101] locfit_1.5-9.6 latticeExtra_0.6-30
## [103] tools_4.2.1 timechange_0.1.1
## [105] future.apply_1.10.0 rstudioapi_0.14
## [107] foreach_1.5.2 foreign_0.8-83
## [109] gridExtra_2.3 prodlim_2019.11.13
## [111] farver_2.1.1 digest_0.6.30
## [113] shiny_1.7.3 lava_1.7.0
## [115] Rcpp_1.0.9 later_1.3.0
## [117] httr_1.4.4 biovizBase_1.44.0
## [119] colorspace_2.1-0 splines_4.2.1
## [121] RBGL_1.72.0 multtest_2.52.0
## [123] plotly_4.10.1 xtable_1.8-4
## [125] jsonlite_1.8.3 futile.options_1.0.1
## [127] timeDate_4021.106 ipred_0.9-13
## [129] R6_2.5.1 Hmisc_4.7-2
## [131] pillar_1.8.1 htmltools_0.5.3
## [133] mime_0.12 glue_1.6.2
## [135] fastmap_1.1.0 BiocParallel_1.30.4
## [137] class_7.3-20 codetools_0.2-18
## [139] utf8_1.2.3 tibble_3.1.8
## [141] curl_4.3.3 zip_2.2.2
## [143] interp_1.1-3 survival_3.4-0
## [145] rmarkdown_2.18 InteractionSet_1.24.0
## [147] munsell_0.5.0 rhdf5_2.40.0
## [149] GenomeInfoDbData_1.2.8 iterators_1.0.14
## [151] HDF5Array_1.24.2 reshape2_1.4.4
## [153] gtable_0.3.1